













Introduction
Risk information published by the US Food and Drug Administration (FDA) plays a critical role in ensuring the safety, effectiveness, and continued performance of approved medical devices. Once a device enters the market, its regulatory oversight does not end. Instead, the FDA continuously monitors device-related risks through structured post-market surveillance systems. Understanding FDA risk information helps manufacturers, regulatory professionals, healthcare providers, and patients make informed decisions while supporting a lifecycle-based approach to medical device safety.
As medical technologies become more complex and widely used, transparent communication of risks associated with FDA approved medical devices has become an essential pillar of patient protection and regulatory compliance.
What Does FDA Approval Mean for Medical Devices?
FDA approval or clearance indicates that a medical device has met defined regulatory requirements for safety and effectiveness based on its intended use. However, the regulatory pathway depends on the device’s risk level and technological novelty.
Most moderate-risk devices enter the US market through the 510(k) clearance pathway, where manufacturers demonstrate substantial equivalence to a legally marketed device. High-risk devices that sustain life or present significant risk require Premarket Approval (PMA), supported by extensive clinical evidence. The De Novo pathway is used for novel devices with no existing predicate but lower to moderate risk.
Regardless of the pathway, FDA authorization does not imply zero risk. Instead, it reflects that identified risks are acceptable when weighed against the device’s clinical benefits.
Understanding FDA Risk Information
FDA risk information is grounded in a structured evaluation of hazards, probabilities, and potential harm. Medical devices are categorized into Class I, II, or III based on their risk profile, intended use, and potential impact on patient health. Class I devices present the lowest risk, while Class III devices carry the highest risk and require the most stringent regulatory oversight.
During premarket review, the FDA assesses device safety and effectiveness using risk–benefit analysis. This evaluation considers whether the clinical benefits outweigh known and foreseeable risks, as well as whether adequate risk control measures are implemented. These principles continue throughout the post-market phase as real-world data becomes available.
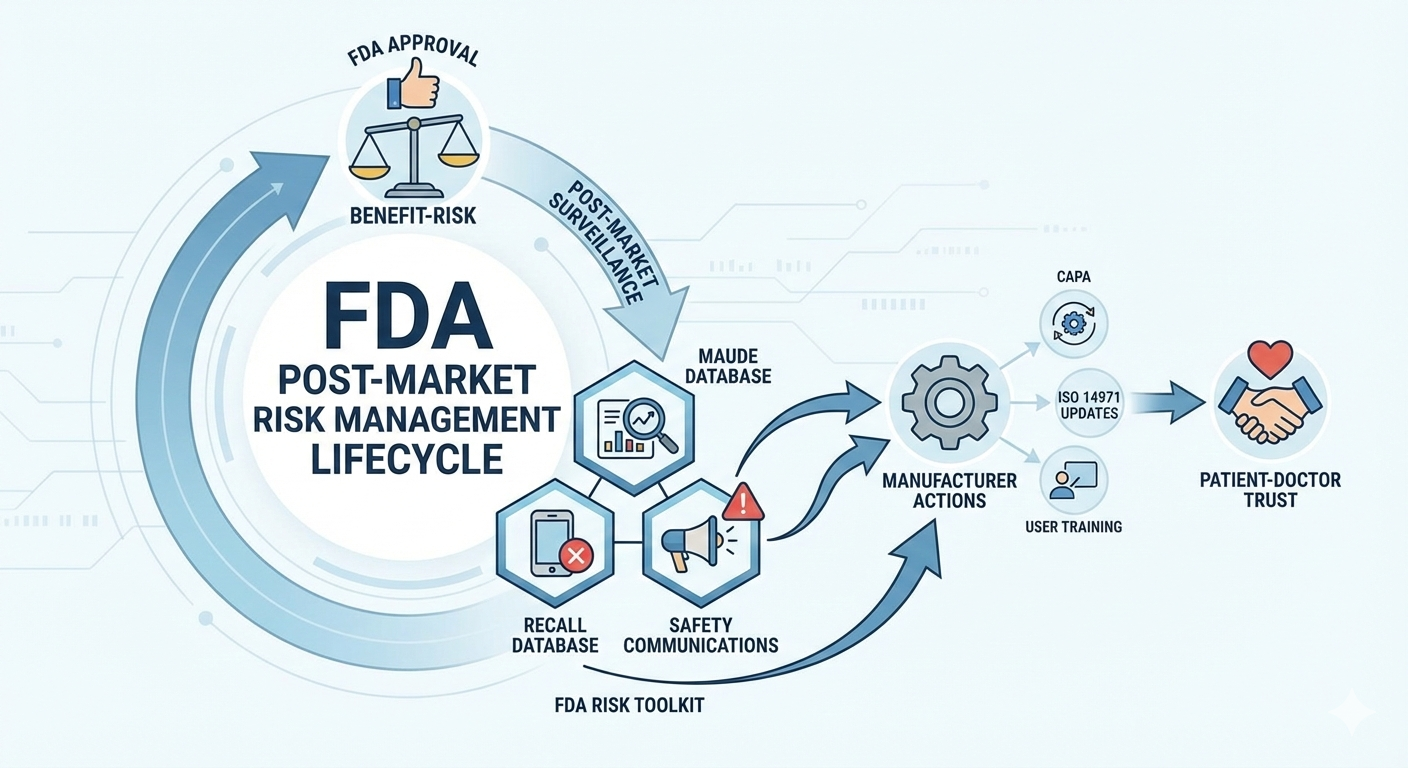
FDA Databases for Risk and Safety Information
The FDA maintains several publicly accessible databases that provide insights into medical device risks and performance after market entry. These systems are essential tools for transparency and continuous safety monitoring.
The Medical Device Recall database lists devices that have been removed or corrected due to safety or performance concerns. Recalls may be voluntary or FDA-mandated and are classified based on severity.
The MAUDE database, or Manufacturer and User Facility Device Experience, collects reports of adverse events, device malfunctions, and serious injuries. While MAUDE data does not establish causality, it helps identify potential safety trends requiring further investigation.
FDA Safety Communications provide timely updates on emerging risks, recommended actions, and device usage precautions. These communications support proactive risk mitigation for healthcare providers and manufacturers.
Together, these systems form the foundation of FDA post-market surveillance and ongoing risk assessment.
Importance of Risk Information for Manufacturers
For manufacturers, FDA risk information is a valuable input for maintaining compliance and improving device performance. Post-market data supports continuous risk management activities, enabling manufacturers to identify new hazards, evaluate residual risks, and refine control measures.
FDA risk data also informs corrective and preventive actions (CAPA), design updates, labeling changes, and user training improvements. Incorporating post-market feedback into the risk management file aligns with FDA expectations and international standards such as ISO 14971.
Proactive monitoring of FDA safety information helps manufacturers avoid regulatory non-compliance, reduce recall risks, and strengthen long-term product reliability.
Impact on Healthcare Providers and Patients
FDA risk information enhances transparency for healthcare providers and patients by supporting informed clinical decisions. Access to recall notices, safety alerts, and adverse event data enables clinicians to evaluate device performance, select appropriate technologies, and manage patient risks effectively.
For patients, this transparency builds trust in the regulatory system and promotes awareness of potential device-related issues. When risk information is communicated clearly and responsibly, it contributes to better health outcomes and patient safety.
Regulatory Responsibilities After FDA Approval
FDA authorization marks the beginning of post-market regulatory responsibilities rather than the end of compliance. Manufacturers are required to maintain ongoing surveillance of their devices, report adverse events, and address identified risks in a timely manner.
Medical Device Reporting (MDR) obligations require manufacturers and user facilities to notify the FDA of serious injuries, malfunctions, and deaths associated with device use. Post-market surveillance studies may also be mandated for certain high-risk devices.
This lifecycle approach reinforces the principle that risk management is a continuous process, integrating premarket analysis with real-world performance data.
Key Takeaways
- FDA approved medical devices are subject to continuous risk monitoring after market entry
- Risk classification and benefit–risk analysis guide FDA regulatory decisions
- Databases such as MAUDE and recall listings support transparency and safety oversight
- Manufacturers use FDA risk information to strengthen compliance and CAPA processes
- Post-market surveillance is essential for protecting patients and maintaining trust
Reference / Further Reading
For a detailed educational overview of how FDA risk information is communicated and interpreted for approved medical devices, readers may refer to resources provided by Operon Strategist at:
Understanding FDA risk information is a shared responsibility across manufacturers, regulators, and healthcare professionals, supporting safer medical technologies and more informed healthcare decisions worldwide.





